
Print Working Directoryの略. current directoryの確認に使用.
$ pwd出力例
/home/cmha # "/" (root)下にある"home" directory配下の"cmha" directory
make directoryの意味. 指定名のdirectoryを作る.
$ mkdir A # "A"という名前のdirectoryを/home/cmha上に作製 (/home/cmha/A). これは相対パス指定の例
# mkdir /home/cmha/A というように絶対パス指定でもOK
# mkdir ~/A とも書ける, "~"はhome directoryに置換される
# 因みに"#"以下は行末までコメントとして無視される
$ mkdir A/B # "A" directory配下に"B" directoryを作製
# mkdir -p A/B という風に-pオプションを使えば
# "A" directoryが存在しなくても両方のdirectoryを一度
# に作製することができる.
Change Directoryの意味. Current directoryの移動に使用.
$ cd A/B $ pwd
/home/cmha/A/B
$ cd .. # ".."は一つ上のdirectoryの意味 $ pwd
/home/cmha/A
$ cd # 引数が無指定の場合はhome directoryに移動 $ pwd
/home/cmha
$ cd - # 引数に"-"を指定すれと直前のcurrent directoryに移動する $ pwd
/home/cmha/A
$ cd ~/A/B # 当然、絶対パスでも指定可能 $ pwd
/home/cmha/A/B
listの意味. あるdirectory内のfileやdirectoryの一覧を表示する.
$ ls ~ # 引数で指定したdirectory(=ここではhome directory)のfile及びdirectoryの一覧表示
$ ls -a ~ # "."から始まるfile・directory名のもの(隠しファイル)も表示
$ ls -l ~ # 詳細表示
$ ls -al ~ # -a オプションと -l オプション の両方を指定した場合
# ls -a -l ~ と書いても同じ
# ls -l -a ~ と書いても同じ
# ls -la ~ と書いても同じ
$ ls # Current directoryのfile・directory一覧表示
file内容を表示する (別画面に遷移する. qとタイプすると終了)
$ less ~/.bashrc
file内容を表示する.
$ cat ~/.bashrc
結果省略
file内容を連結(Catenation)表示する
$ echo aaa > f1 # "aaa"という内容のファイルf1を作製
$ echo bbb > f2 # "bbb"という内容のファイルf2を作製
$ cat f1 f2 > f3 # f1 f2の順でファイル内容を連結した
# f3を作製
$ cat f3
aaa bbb aaa
Copyの意味. file・directoryを複製する.
$ mkdir C $ cp f1 C/f4 # f1のコピーをf1という名前でC配下に作製 $ cat C/f4
aaa
$ cp -r C D # directoryをコピーするときは-rオプションが必要 $ cat C/f4
aaa
moveの意味.mv from toの書式.toが既存のdirectoryの場合、fromをtoの中に移動する.
$ mv f3 C $ ls
C D f1 f2
$ ls C
f3 f4
ファイル名を変更する
$ mv f2 f_2 $ ls
f1 f_2 C D
$ mv f1 f_2 # 移動先に既存のファイル名を指定した場合 $ cat f_2
aaa
removeの意味. file・directoryの削除.
$ rm f_2 $ ls
C D
$ rm -r C # directoryを削除する際は-r オプションをつける. $ ls
D
$ vim hoge # "hoge"という名前のファイルをvimで編集する.
今回はVIMというTEXT editorを用いてファイルを編集する. VIMにはノーマルモードとインサートモードがある(他のモードも多くあるがここでは省略). 下表を参考にしながら(1)ノーマルモードからインサートモードに移行、 (2) 適当に文字を入力、 (3) 再度ノーマルモードに戻り、 作業内容をファイルに保存して終了するまでの作業を行う.
| i | インサートモードに移行する |
| d | 削除 |
| dd | 行削除 |
| y | コピー |
| v | 領域選択 |
| V | 行選択 |
| yy | 行コピー |
| p | 貼り付け |
| u | 元に戻す |
| :w | ファイルに(上書き)保存 |
| :wq | ファイルに(上書き)保存して終了 |
| :q! | ファイルに変更点を保存せずに強制終了 |
| ESC | ノーマルモードに移行 |
command実行結果の出力先を標準出力からFILEにリダイレクト
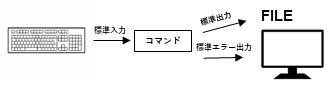
$ echo atgc
atgc
$ echo atgc > hoge # コマンドの出力結果を">"を用いて画面表示からファイルにリダイレクト $ cat hoge
atgc
パイプ"|"を用いてechoコマンドの出力結果をrevコマンドの入力としている.
$ echo atgc | rev # パイプ"|"を用いてechoコマンドの出力結果をrevコマンドの入力としている.
# revコマンドは行内の文字列を逆順で出力するコマンド
cgta
$ x=atgc # "atgc"という文字列を変数xに代入."="の前後に空白を含まぬよう!! $ echo $x # 変数xの内容を参照する際は$xとする
atgc
$ y=$(echo $x | rev) # コマンドで実行結果を変数に代入 $ echo REVSEQ is $y # 固定文章中に変数を埋め込んでいる例
REVSEQ is cgta
trコマンドは文字変換・文字削除を行う.
$ echo atgc | tr 'g' 'G' # gをGに変換する
atGc
$ echo atgc | tr 'a-z' 'A-Z' # 小文字を大文字に変換する
ATGC
$ echo atacagg | tr -d 'at' # aとtを除く. (GC比算出の際などで利用)
cgg
以下のような不統一な形式のデータd1は 簡単に一貫した形式に整理できる
$ cat d1 # 管理の悪いプライマー表
ata gcc atc ggg ata gca ac GCACATGCCAGTACCATG catgcagtatgcttactcgat GCT ACA GCC ATT GCA GGG ATC aatggtACGTAGCAGTccagta
$ cat d1 | tr 'a-z' 'A-Z' | tr -d ' ' > tmp # あとで使うためtmpファイルに保存 $ cat tmp
ATAGCCATCGGGATAGCAAC GCACATGCCAGTACCATG CATGCAGTATGCTTACTCGAT GCTACAGCCATTGCAGGGATC AATGGTACGTAGCAGTCCAGTA
プライマーの総数を知りたい場合は行数を数えればよい. 行数を知るにはwcコマンドに-lオプションは付与して実行する.
$ cat tmp | wc -l
5
次の課題としてtmpファイル中の各プライマーの塩基長を求めてみる. 塩基長、すなわち文字列長はwcコマンドに -mオプションを付与することで出力できる. まずコマンドの挙動を簡単な例で確認してみる.
$ echo atgc | wc -m # -m: 文字数出力
5 # echoコマンドの出力は行末に改行文字(\n)を含むため +1
$ echo -n atgc | wc -m # (解決策1): -n オプションを指定して行末の改行抑制
4
$ echo atgc | tr -d '\n' | wc -m # (解決策2): trコマンドで改行文字を除く
4
さてコマンドの挙動は分かったので先ほど作成したtmpデータの各行の塩基長を求めてみる.
$ cat tmp | wc -m # これでは各行の塩基長でなく全塩基長が算出される.
107
行ごとに処理を実行したい場合はwhile read文を使用するとよい.
$ cat tmp | while read i; do echo -n $i | wc -m ; done
20 18 21 21 22
tmpデータは一列のデータであるため、変数を1つ分(上記ではi [変数名は任意])用意した. 複数列のデータd2の場合には以下のように 3つ分の変数(i, j, k)を用意するとよい.
$ cat d2
2022 1 1 2022 4 1 2022 5 15 2022 8 23
$ cat d2 | while read i j k; do echo $k/$j/$i ; done
1/1/2022 1/4/2022 15/5/2022 23/8/2022
次にデータの抽出や並び替えに関する課題について考えてみる. データ抽出の課題の多くは表形式[=矩形]データから目的の行や列を取り出すことを意味する. 目的の行や列は文字列のマッチングや比較演算,または行番号・列番号を指定することで取り出す. 正規表現を学習すると複雑な文字列マッチング条件を指定することも可能だが今回はほとんど取り扱わない. データ抽出を行うコマンドにはgrepやawkがある. データd3を用いて実習してみよう.
$ cat d3 | head # データ構造の確認にはheadコマンドで行頭のみ出力するとよい. # 1:遺伝子名 2:スコア値 3:p値
Gnai3 1.22589 8.4791e-05 Cdc45 1.09439 0.434275 Scml2 0.898889 0.675262 Narf 3.68168 2.60196e-23 Cav2 1.17894 0.85503 Klf6 1.82039 1.54732e-05 Scmh1 1.38563 0.0373061 Cox5a 1.04905 0.620365 Ngfr 0.786942 0.365366 Wnt9a 1.95754 0.211958
$ cat d3 | grep Fgf # Fgfという語を含む行のみ抽出
Fgfr4 0.88575 0.644229 Fgf22 1.28104 0.634742 Fgf9 1.26628 0.162239 Fgf12 1.30162 0.0543288 Fgf14 1.06723 0.81295 Fgfr2 0.95496 0.897533 Fgfr1 1.06513 0.911698 Fgf1 1.0326 0.919811 Fgfr1op2 1.09272 0.4113 Fgfbp3 1.24051 0.604392 Fgfr3 1.44692 0.495306 Fgfr1op 0.838637 0.096374
$ cat d3 | grep Fgf | grep -v Fgfr # -v: Fgfrを除外
Fgf22 1.28104 0.634742 Fgf9 1.26628 0.162239 Fgf12 1.30162 0.0543288 Fgf14 1.06723 0.81295 Fgf1 1.0326 0.919811 Fgfbp3 1.24051 0.604392
遺伝子リストd4記載の遺伝子のみ抽出するには -f list_fileのように指定する.
$ cat d4 # 抽出したい遺伝子エントリーのリスト
Hnf4a Bnip3l Bhlhe40 Nkx6-1 Foxo3
$ cat d3 | grep -f d4
Hnf4a 1.10896 0.604998 Bnip3l 3.8967 3.55386e-64 Bhlhe40 6.71858 1.81051e-91 Nkx6-1 1.04578 0.402985 Foxo3 1.99961 7.32318e-18
次にawkコマンドを用いた場合について考えてみる. awkを使えばgrepのような文字列のマッチングだけでなく、比較演算を利用したデータ抽出ができる.
| < | より小さい |
| <= | より小さい又は等しい |
| < | less than |
| == | 同じ |
| != | 同じでない |
| > | より大きい |
| => | より大きい又は等しい |
| ~ | パターンにマッチする |
| !~ | パターンにマッチしない |
awkは以下のような書式で使用し、以下のように挙動する(デフォルト時).
$ awk 'パターン1{アクション1} パターン2{アクション2} ....'
cat d3 | awk '$2 > 10 {print $1, $2, $3}' # Scoreが10より大きいものだけ抽出 (比較演算).
# パターンは $2 > 10
# 2列目が10より大きいもののみアクションを実行する.
# アクションは{print $1, $2, $3}
# 1列目($1), 2列目($2), 3列目($3)を出力するという意味.
# {print $1, $2, $3}のように全列名を指定するのは面倒だ.
# $0は全列($1, $2, $3)を意味する変数のため,アクションの部分は{print $0}としてもよい.
# アクションの部分を省略した場合は全列出力の意味となるため、awk '$2 > 10'とするだけで十分.
(見やすさのため出力結果を整形している) Ak4 16.8428 5.01187e-44 Adm 11.3649 1.34586e-43 Lzts1 11.0648 0.0871485 Kcnh3 42.8431 4.89463e-06 Bnip3 10.5844 1 Hspa1b 14.144 1.48252e-169 Hspa1a 12.5628 8.97429e-152 Morf4l1-ps1 12.9427 0.519003
$ cat d3 | awk '$1=="Fgf1"'
# 1列目が完全にFgf1という文字列と一致する行を出力する.
# このように文字列は二重引用符"で囲む.
Fgf1 1.0326 0.919811
$ cat d3 | awk '$1 ~ "Fgf"' # 1列目がFgfという文字列を含む行を出力する.
Fgfr4 0.88575 0.644229 Fgfr2 0.95496 0.897533 Fgfr1 1.06513 0.911698 Fgfr1op2 1.09272 0.4113 Fgfr3 1.44692 0.495306 Fgfr1op 0.838637 0.096374
次にデータの並び替えについて考えてみよう. データの並び替えにはsortコマンドを用いる.
$ cat d3 | sort | head # 1列目を最優先キー列として昇順で並び替え.
A1cf 1.0505 0.863402 A2ml1 1.16054 0.735985 AA414768 0.997071 0.993518 AA465934 0.927646 0.878384 AA467197 1.51277 0.367104 AA474408 0.866833 0.66265 AA986860 1.31109 0.582374 AI413582 1.31488 0.0263645 AI429214 1.22011 0.699422 AI467606 1.05248 0.948288
$ cat d3 | sort -k 2n,2 | head
# -k 2,2とすれば2列目を最優先キー列として並び替え.
# 2列目は数値なのでさらにnオプションを付与している.
Tmem254c 0.00352429 4.09562e-09 Lgals3 0.0572853 0.0503072 Zfp991 0.0801197 0.0325747 Cachd1 0.12652 0.00188426 Adam4 0.146645 0.00157841 Nphp3 0.203929 0.00639455 Rpl9-ps7 0.236934 0.151531 Ndnf 0.260776 9.82969e-05 Hecw2 0.27597 0.0162911 Kcnq5 0.276302 0.0423721
$ cat d3 | sort -k 2nr,2 | head # さらにrオプションを付与すれば降順に並び替える
Kcnh3 42.8431 4.89463e-06 Ak4 16.8428 5.01187e-44 Hspa1b 14.144 1.48252e-169 Morf4l1-ps1 12.9427 0.519003 Hspa1a 12.5628 8.97429e-152 Adm 11.3649 1.34586e-43 Lzts1 11.0648 0.0871485 Bnip3 10.5844 1 Ankrd37 9.29321 5.60919e-44 Muc2 9.28706 8.59271e-07
$ cat d3 | sort -k 3g,3 | head # 数値が指数表記を含む場合はnの代わりにgオプションを付与する
Pgam1 2.96626 2.00447e-265 Gpi1 2.69398 1.15611e-260 Pgk1 2.76836 4.0738e-249 Pfkl 3.99423 2.26464e-239 Eno1 3.43571 2.6485e-200 Slc2a1 4.55824 9.50605e-198 Tpi1 3.07655 6.99842e-197 Hspa1b 14.144 1.48252e-169 Slc16a3 7.09728 5.78096e-154 Hspa1a 12.5628 8.97429e-152
以上を応用して実践的な課題を考えてみる.
(課題1) 有意(p値<0.05)でかつfold change(=スコア)が2以上の遺伝子はいくつあるか?
$ cat d3 | awk '$3 < 0.05' | awk '$2 ≥ 2' | wc -l
135
(課題2) 有意でかつfold changeが上位5番目までの遺伝子を抽出せよ.
$ cat d3 | awk '$3 < 0.05' | sort -k 2nr,2 | head -n 5 # head -n 5で行頭より5行目までを抽出.
Kcnh3 42.8431 4.89463e-06 Ak4 16.8428 5.01187e-44 Hspa1b 14.144 1.48252e-169 Hspa1a 12.5628 8.97429e-152 Adm 11.3649 1.34586e-43
コマンドを多用する処理内容などはファイルに処理内容を記述後にshコマンドで実行するとよい. 処理内容を記述したファイルをシェルスクリプト(「台本」という意味)という. シェルスクリプトの記述法は自由度が高いため、適宜スペースや改行などを加えて読みやすく工夫することができる. 例えば以下のようにbaseという名のスクリプトファイルを作成してみる.
$ vim base
cat d1 | # 入力データ
tr -dc 'atgcATGC\n' | # 塩基配列文字と改行以外を除く
tr 'atgc' 'ATGC' | # 大文字に統一
while read seq ; do
revcomp=$(
echo $seq | # 元の塩基配列
rev | # 逆順変換
tr 'ATGC' 'TACG' ) # 相補関係に変換
N_total=$(echo -n $seq | wc -m) # 全塩基長
N_gc=$( echo -n $seq | tr -d 'AT' | wc -m) # GC配列長
echo $seq $revcomp $N_total $N_gc
done
次にこのシェルスクリプトをデータd1を処理するためだけでなく、 より一般的なコマンドとして今後利用していくことを考える. コマンドは以下の手順で作成・利用することができる.
$ vim base
#!/bin/sh
cat $1 | # 入力データ
tr -dc 'atgcATGC\n' | # 塩基配列文字と改行以外を除く
tr 'atgc' 'ATGC' | # 大文字に統一
while read seq ; do
revcomp=$(
echo $seq | # 元の塩基配列
rev | # 逆順変換
tr 'ATGC' 'TACG' ) # 相補関係に変換
N_total=$(echo -n $seq | wc -m) # 全塩基長
N_gc=$( echo -n $seq | tr -d 'AT' | wc -m) # GC配列長
echo $seq $revcomp $N_total $N_gc
done
コマンドとして利用していくために先ほどのスクリプトから2か所変更した. 1つ目は文頭に#!/bin/sh (=シバンという)を追加することで、このスクリプトを/bin/shで処理するよう明示した. 2つ目は入力データをコマンドの引数として指定できるように、入力データの箇所を変数$1に置換した. $1は位置パラメータといい、このコマンドに指定した最初の引数の値が代入される. 位置パラメータは$1, $2, $3....といように複数指定することができる.
コマンドとして実行可能であるためにはスクリプトファイルに実行権限を与える必要がある.
$ chmod +x base # "chomd +x"によりコマンドとして実行可能なファイルとなる.
$ echo $PATH # PATHは組込変数
# ":"区切り.コマンド用directoryへのパスの一覧
# これらdirectory内ファイル名はそのままコマンド名として認識される.
/usr/local/sbin:/usr/local/bin:/usr/sbin:/usr/bin:/sbin
$ vim ~/.profile # PATH変数は通常~/.profile内で定義されている.
ファイル内のPATHの記載部分を PATH=~/bin:/usr/local/sbin:/usr/local/bin:/usr/sbin:/usr/bin:/sbin などのように書き換えて 自分の作ったコマンドを入れるdirectory~/binをPATHの先頭に追加.
$ . ~/.profile # "."コマンドを用いて変更内容を反映させる. $ echo $PATH
/home/tom/bin:/usr/local/sbin:/usr/local/bin:/usr/sbin:/usr/bin:/sbin
$ mkdir ~/bin # 存在しないなら作製.
$ mv base ~/bin # scriptをPATHの通った~/binに移動.
# これでコマンドとして使用できるはずだから...
$ base d1 # 確認!!
結果省略
curlコマンドを利用すればネットワーク越しにファイルを取得できる. コマンドの使用形式は以下の通りである.
$ curl -O URL # リクエスト先のファイル名で出力. $ curl -o filename URL # 指定したファイル名で出力.GENCODEからversion30のマウスの遺伝子アノテーションデータをダウンロードする.
$ curl -O http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M30/gencode.vM30.annotation.gtf.gz
gzという拡張子(*.gz)がついてるファイルは通常gzipで圧縮処理されたファイルである.
ファイルサイズの確認
$ ls -l gencode.vM30.annotation.gtf.gz
-rw-r--r-- 1 tom tom 29248040 10月 9 21:35 gencode.vM30.annotation.gtf.gz
ファイルサイズの確認(より分かりやすい表示)
$ ls -lh gencode.vM30.annotation.gtf.gz
-rw-r--r-- 1 tom tom 28M 10月 9 21:35 gencode.vM30.annotation.gtf.gz
ファイル内容の表示
$ cat gencode.vM30.annotation.gtf.gz | head # 圧縮ファイルなのでよく分からない.
圧縮ファイルを解凍
$ gunzip gencode.vM30.annotation.gtf.gz
ファイルサイズの確認
$ ls -lh gencode.vM30.annotation.gtf
-rw-r--r-- 1 tom tom 868M 10月 9 21:35 gencode.vM30.annotation.gtf
ファイル内容の表示
$ cat gencode.vM30.annotation.gtf | head # ちゃんと読めることを確認.
ファイルの圧縮
$ gzip gencode.vM30.annotation.gtf
zcatコマンドを使用すればファイルを圧縮状態のまま、非圧縮状態の内容を標準出力することができる.
$ zcat gencode.vM30.annotation.gtf.gz | head # zcatが利用できない場合はgunzip -c
複数のファイルやディレクトリをまとめたファイルをアーカイブ[=書庫]という. ZIPアーカイブを見たことがあると思うが、Unixの世界ではtarアーカイブを伝統的に使用する. tarアーカイブファイルはtarコマンドを用いて操作する.
dirのアーカイブout.tarを作成.
$ tar cf out.tar dir
# cオプションはcreate, fオプションはfileの意味であり、
# 伝統的に-を付けずにオプション指定する.
dirのgzip圧縮アーカイブout.tar.gzを作成.
$ tar czf out.tar.gz dir # zオプションを付けている.gzip圧縮アーカイブout.tarを展開する.
$ tar xzf out.tar.gz #xオプションはextractの意味である.
前章でダウンロードしてきたgencode.vM30.annotation.gtf.gzファイルを用いてGTFファイルの中身を探索してみる. Gencodeの GTF形式の仕様や 統計処理データ の詳細が公開されているために矩形データ処理の練習題材としては好適である.
まずはheadコマンドを用いてデータ構造の概要を確認しよう.
$ zcat gencode.vM30.annotation.gtf.gz | head
##description: evidence-based annotation of the mouse genome (GRCm39), version M30 (Ensembl 107) ##provider: GENCODE ##contact: gencode-help@ebi.ac.uk ##format: gtf ##date: 2022-05-12 chr1 HAVANA gene 3143476 3144545 . + . gene_id "ENSMUSG00000102693.2"; gene_type "TEC"; gene_name "4933401J01Rik"; level 2; mgi_id "MGI:1918292"; havana_gene "OTTMUSG00000049935.1"; chr1 HAVANA transcript 3143476 3144545 . + . gene_id "ENSMUSG00000102693.2"; transcript_id "ENSMUST00000193812.2"; gene_type "TEC"; gene_name "4933401J01Rik"; transcript_type "TEC"; transcript_name "4933401J01Rik-201"; level 2; transcript_support_level "NA"; mgi_id "MGI:1918292"; tag "basic"; tag "Ensembl_canonical"; havana_gene "OTTMUSG00000049935.1"; havana_transcript "OTTMUST00000127109.1"; chr1 HAVANA exon 3143476 3144545 . + . gene_id "ENSMUSG00000102693.2"; transcript_id "ENSMUST00000193812.2"; gene_type "TEC"; gene_name "4933401J01Rik"; transcript_type "TEC"; transcript_name "4933401J01Rik-201"; exon_number 1; exon_id "ENSMUSE00001343744.2"; level 2; transcript_support_level "NA"; mgi_id "MGI:1918292"; tag "basic"; tag "Ensembl_canonical"; havana_gene "OTTMUSG00000049935.1"; havana_transcript "OTTMUST00000127109.1"; chr1 ENSEMBL gene 3172239 3172348 . + . gene_id "ENSMUSG00000064842.3"; gene_type "snRNA"; gene_name "Gm26206"; level 3; mgi_id "MGI:5455983"; chr1 ENSEMBL transcript 3172239 3172348 . + . gene_id "ENSMUSG00000064842.3"; transcript_id "ENSMUST00000082908.3"; gene_type "snRNA"; gene_name "Gm26206"; transcript_type "snRNA"; transcript_name "Gm26206-201"; level 3; transcript_support_level "NA"; mgi_id "MGI:5455983"; tag "basic"; tag "Ensembl_canonical";
##からはじまるコメント行の後にタブ区切り・行指向の矩形データであることが分かる. そこでまずコメント行を除いた後、1行目だけを取り出し、各列を行ごとに変換することで、各列のデータ種類を確認する.
$ zcat gencode.vM30.annotation.gtf.gz | grep -v '^#' | head -n 1 |
tr '\t' '\n' | awk '{print NR, $0}'
# grep -v '^#' で文頭にあるコメント行を除いている.
# ''又は""で囲むことで#のコメントとしての意味を失わせることができる.
# tr '\t' '\n'はタブ(\t)を改行(\n)に変換している.
# awk '{print NR, $0}' で行頭に行番号を附している.
1 chr1 2 HAVANA 3 gene 4 3143476 5 3144545 6 . 7 + 8 . 9 gene_id "ENSMUSG00000102693.2"; gene_type "TEC"; gene_name "4933401J01Rik"; level 2; mgi_id "MGI:1918292"; havana_gene "OTTMUSG00000049935.1";
3列目がgeneのデータを取得すれば全遺伝子の情報が取得できる. 抽出後の行数を算出し、 WEB上で公開されているSTATSデータと照合してみる.
$ zcat gencode.vM30.annotation.gtf.gz | grep -v '^#' | awk -F'\t' '$3=="gene"' | wc -l # awk -F '\t' によりデフォルトのスペース・タブ区切りでなくタブ区切りのみであることを指定
56691
全遺伝子情報が取得できていることが確認出来たら、次にENSEMBL GENE IDと遺伝子シンボル名との対応表を作成してみよう.
$ zcat gencode.vM30.annotation.gtf.gz | grep -v '^#' | awk -F'\t' '$3=="gene"' |
awk -F '\t' '{print $9}' | awk -F ';' '{print $1, $3}' | awk '{print $2, $4}' |
tr -d '"' | sort | head
# GTFファイルの9列目に目的のENSEMBL IDとGene Symbleのデータがある.
ENSMUSG00000000001.5 Gnai3 ENSMUSG00000000003.16 Pbsn ENSMUSG00000000028.16 Cdc45 ENSMUSG00000000031.18 H19 ENSMUSG00000000037.18 Scml2 ENSMUSG00000000049.12 Apoh ENSMUSG00000000056.8 Narf ENSMUSG00000000058.7 Cav2 ENSMUSG00000000078.8 Klf6 ENSMUSG00000000085.17 Scmh1