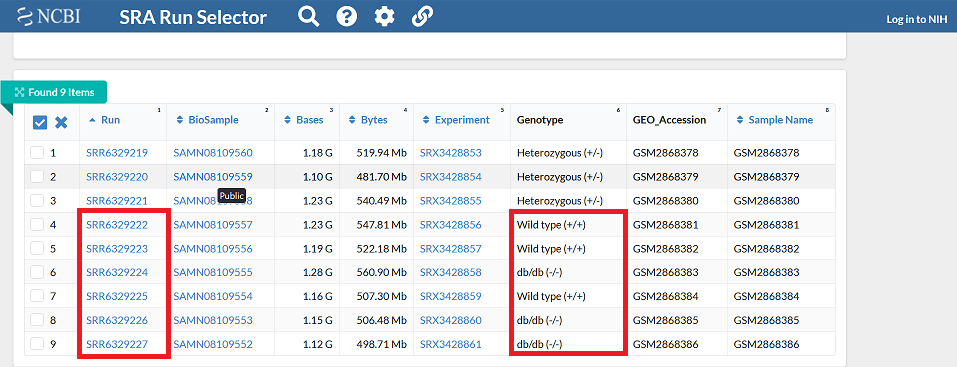
野生型マウスと2型糖尿病マウス(db/db)の膵島における遺伝子発現を 比較した研究( PMID:29542001)データを使用する. 論文上の記載からGEOリポジトリにID番号:GSE107489で登録されていることがわかる. GSE107489@GEOから解析済みデータが入手できるが今回は解析前の生データから再解析する. そこでSRA Run Selectorをクリックし、SRAデータベースのページ(ID:PRJNA420351)に遷移する. Run IDを確認する. Accession ListをダウンロードすればID一覧を得ることができる.
まずは以下のようなプロジェクト専用のdirectory構造を準備しよう. 今回はすでに準備済みのGSE107489 tarballを ダウンロード・解凍して使用する.
$ tar xf GSE107489.tar # tarballの解凍
GSE107489 ├── SCRIPT # スクリプトファイルの配置directory │ ├── download_from_gencode.sh # Reference dataのダウンロードスクリプト │ ├── make_gid2name.sh # 遺伝子IDと遺伝子名の対応表作成スクリプト │ ├── make_tx2gene.sh # 転写産物IDと遺伝子IDの対応表作成スクリプト │ ├── sra.sh # 生データ入手するためのスクリプト │ ├── trim.sh # トリミング実行スクリプト │ ├── map_index.sh # マッピングインデックス作成スクリプト │ ├── map.sh # マッピング実行スクリプト │ ├── deg.ipynb # 発現差解析スクリプト │ └── postdeg.sh # 発現差解析データ整形スクリプト ├── REFERENCE # download_from_gencode.sh, make_gid2name.sh, make_tx2gene実行結果を配置するdirectory ├── SOURCE # sra.sh実行結果を配置するdirectory ├── TRIM # trim.sh実行結果を配置するdirectory ├── INDEX # map_index.sh実行結果を配置するdirectory ├── MAP # map.sh実行結果を配置するdirectory ├── DEG # deg.sh, postdeg.sh実行結果を配置するdirectory └── sample_info # 解析対象の試料情報を記述したファイル.
sample_infoの内容は以下の通りである.
SRR6329222 wt_1 wt SRR6329223 wt_2 wt SRR6329225 wt_3 wt SRR6329224 db_1 db SRR6329226 db_2 db SRR6329227 db_3 db
以下のようにRNA-seq解析に必要なツールを導入する.
$ conda create -n sra # sra環境を作る. $ conda create -n trim # trim環境を作る. $ conda create -n map # map環境を作る. $ conda create -n deg # deg環境を作る. $ conda info -e # 環境の一覧を表示し、確認.
$ conda activate sra # sra環境に入る. $ conda install -c bioconda sra-tools # sra-tools"をインストール. $ conda activate trim # trim環境に入る. $ conda install -c bioconda trim-galore # trim-galore"をインストール. $ conda activate map # map環境に入る. $ conda install -c bioconda salmon # salmonをインストール. $ conda activate deg # deg環境に入る. $ conda install -c bioconda bioconductor-deseq2 bioconductor-tximport # DESEQ2とtximportをインストール. $ conda install -c conda-forge jupyterlab r-irkernel # Jupyter利用に必要なもの.
解析を実行する前にPCの論理コア(スレッド)数の確認しておく. 以下の例では最大8スレッドまで指定できる.
$ cat /proc/cpuinfo | grep processor | wc -l # WSL2 user $ sysctl -n hw.logicalcpu_max # macOS user
8
SRAデータベースから生データを入手するためのスクリプトsra.sh の内容は以下の通りである.
# このスクリプトはProject directory上からsh SCRIPT/sra.shのように
# 実行することを想定している.
info=sample_info
thread=8
tmp_dir=/tmp
output_dir=SOURCE
cat $info |
while read i j k ; do
fasterq-dump $i \
-t $tmp_dir \
-O $output_dir \
-e $thread \
-p
gzip $output_dir/$i.fastq
done
解析(データのダウンロードとGZIP圧縮)実行
$ conda activate sra $ sh SCRIPT/sra.sh
GENCODEからReferenceデータを入手するためのスクリプト download_from_gencode.shの内容は以下の通りである.
# このスクリプトはProject directory上からsh SCRIPT/download_from_gencode.shのように # 実行することを想定している. MUS=30 HS=41 URL_M=http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse URL_H=http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human cd REFERENCE # (必要なら) DOWNLOADしたファイルを配置するDirectoryに移動 # GTF curl -O $URL_M/release_M$MUS/gencode.vM$MUS.annotation.gtf.gz #curl -O $URL_H/release_$HS/gencode.v$HS.annotation.gtf.gz # TRANSCRIPTOME curl -O $URL_M/release_M$MUS/gencode.vM$MUS.transcripts.fa.gz #curl -O $URL_H/release_$HS/gencode.v$HS.transcripts.fa.gz # GENOME #curl -O $URL_M/release_M$MUS/GRCm39.primary_assembly.genome.fa.gz #curl -O $URL_H/release_$HS/GRCh38.primary_assembly.genome.fa.gz
downloadの実行
$ sh SCRIPT/download_from_gencode.sh
Transcription IDとGene IDの対応表tx2geneを作成するスクリプト make_tx2gene.shの内容は以下の通りである.
# このスクリプトはProject directory上からsh SCRIPT/make_tx2gene.shのように
# 実行することを想定している.
input_file=gencode.vM30.annotation.gtf.gz
output_file=tx2gene
cd REFERENCE # (必要なら)事前に*.gtfファイルの配置されたdirectoryに移動しておく.
zcat $input_file | # zcatが使用できぬ場合はgunzip -c
grep -v '^#' | # 冒頭のコメント行を除く処理
awk -F'\t' '$3=="transcript"' | # 3列目がtranscriptであるエントリーのみ抽出
awk -F'\t' '{print $9}' | # 9列目に遺伝子IDと遺伝子名の情報があるので抽出
awk -F ';' '{print $2, $1}' | # ;区切りの2,1列目の順に抽出
awk '{print $2, $4}' | # スペース区切りの2,4列目を抽出
tr -d '"' | # 不要な引用符を除く
tr '.' ' ' | # 以下2行はIDのバージョン番号を取除くための処理
awk '{print $1, $3}' | # 正規表現で正確にマッチさせ置換する処理の方が望ましい
sort < $output_file
Gene IDと遺伝子名の対応表gid2nameを作成するスクリプト make_gid2name.shの内容は以下の通りである.
# このスクリプトはProject directory上からsh SCRIPT/make_gid2name.shのように
# 実行することを想定している.
input_file=gencode.vM30.annotation.gtf.gz
output_file=gid2name
cd REFERENCE # (必要なら)事前に*.gtfファイルの配置されたdirectoryに移動しておく.
zcat $input_file | # zcatが使用できぬ場合はgunzip -c
grep -v '^#' | # 冒頭のコメント行を除く処理
awk -F'\t' '$3=="gene"' | # 3列目がgeneであるエントリーのみ抽出
awk -F'\t' '{print $9}' | # 9列目に遺伝子IDと遺伝子名の情報があるので抽出
awk -F ';' '{print $1, $3}' | # ;区切りの1,3列目を抽出
awk '{print $2, $4}' | # スペース区切りの2,4列目を抽出
tr -d '"' | # 不要な引用符を除く
tr '.' ' ' | # 以下2行はGene IDのバージョン番号を取除くための処理
awk '{print $1, $3}' | # 正規表現で正確にマッチさせ置換する処理の方が望ましい
sort < $output_file
ID対応表の作成実行
$ sh SCRIPT/make_tx2gene.sh $ sh SCRIPT/make_gid2name.sh
trim.shスクリプト内容は以下の通りである.
info=sample_info
input_dir=SOURCE
output_dir=TRIM
cat $info |
while read i j k; do
trim_galore \
-o $output_dir \
$input_dir/$i.fastq.gz
done
解析(トリミング)実行.
$ conda activate trim $ sh SCRIPT/trim.sh
Mappingのためのindex(再利用可)を作成する. map_index.shスクリプト内容は以下の通りである.
thread=8 ref_transcripts=REFERENCE/gencode.vM30.transcripts.fa.gz output_dir=INDEX salmon index \ -p $thread \ -t $ref_transcripts \ -i $output_dir \ -k 31 \ --gencode
解析(index作成)の実行.
$ conda activate map $ sh SCRIPT/map_index.sh
map.shスクリプト内容は以下の通りである.
info=sample_info
thread=8
input_dir=TRIM
index_dir=INDEX
output_dir=MAP
cat $info |
while read i j k ; do
salmon quant \
-l A \
-i $index_dir \
-r $input_dir/${i}_trimmed.fq.gz \
-p $thread \
--validateMappings
-o $output_dir/$i
done
解析(mapping)実行
$ sh SCRIPT/map.sh
DEG解析のためのRスクリプトdeg.ipynbをJupyter環境から実行する.
$ conda activate deg $ jupyter lab SCRIPT/deg.ipynb
# load
library("tximport")
library("jsonlite")
library("DESeq2")
setwd("..") # GSE107489/SCRIPTからGSE107489へ作業directoryを移動
# set parameters
info_path <- "sample_info"
tx2gene <- "tx2gene"
map_dir <- "MAP"
ref_dir <- "REFERENCE"
deg_dir <- "DEG"
output_txi <- "txi"
output_d2 <- "deseq2"
output_pca <- "pca.pdf"
output_heatmap <- "heatmap.pdf"
# sample information
info <- read.table(info_path, header = FALSE, sep = " ")
info$V3 <- factor(info$V3, levels = c("wt","db"))
path <- file.path(map_dir, info$V1, "quant.sf")
names(path) <- info$V2
tx2gene <- read.table(file.path(ref_dir, tx2gene), header = FALSE, sep = " ")
# tximport
txi <- tximport(path,
type = "salmon",
tx2gene = tx2gene,
ignoreTxVersion = TRUE )
# DESeq2
dds <- DESeqDataSetFromTximport(txi, info, ~ V3)
dds <- DESeq(dds)
res <- results(dds)
rld <- rlog(dds)
rld_mat <- assay(rld)
rld_cor <- cor(rld_mat)
# output data
write.table(txi, file.path(deg_dir, output_txi), append = FALSE)
write.table(res, file.path(deg_dir, output_d2), append = FALSE)
pdf(file.path(deg_dir, output_pca))
plotPCA(rld, intgroup = "V3")
dev.off()
pdf(file.path(deg_dir, output_heatmap))
heatmap(rld_cor)
dev.off()
DEGs解析データを整形するためのpostdeg.shの内容は以下の通りである.
input=DEG/deseq2
output=DEG/deseq2_processed.csv
gid2name=REFERENCE/gid2name
cat $input |
# ヘッダーを除く
tail -n +2 |
# 1:ID 3:logFC 7:adjusted_p_val
awk '{print $1, $3, $7}' |
# 引用符を除く
tr -d '"' |
# 欠損値を含む行を除く
awk '$3!="NA"' |
awk '$4!="NA"' |
# 2:logFCを2:FCに変換する
awk '{print $1, 2^$2, $3}' |
# 遺伝子名情報を内部結合で追加する
join -o 1.1 2.2 1.2 1.3 - $gid2name |
# 1番目のファイル: d2_processed
# 2番目のファイル: gid2name
#
# 1列目を共通キーとして両者を内部結合している.
# キー列(1列目)はあらかじめソートされている必要がある.
#
# -o 1.1 2.2 1.2 1.3 の箇所で何をどの順に出力するかを指定.
# 1.1: ファイル1の1列目
# 2.2: ファイル2の2列目
# 1.2: ファイル1の2列目
# 1.3: ファイル1の3列目
# EXCELで開きやすいようCSV形式にする
tr ' ' ',' < $output