
ChIP-seqデータ解析手順の大略を示す。
シーケンス深度を十分に確保しないと峰検出がうまくいかないことは容易に想像できるであろう。 シーケンス深度がどのくらい必要とされるかは対照生物のゲノムサイズと対象タンパクの結合部位サイズに依存する。 ENCODEプロジェクトのデータ基準値に従えば、解析対象が マウス・ヒトの転写因子では20Mリード(またはリード対、以下同様)、 マウス・ヒトのヒストンでは Narrow Marks (H2AFZ, H3ac, H3K27ac, H3K4me2, H3K4me3, H3K9ac) の場合には20Mリード、 Broad Marks (H3F3A, H3K27me3, H3K36me3, H3K4me1, H3K79me2, H3K79me3, H3K9me1, H3K9me2, H4K20me1)やH3K9me3の場合には45Mリードを確保する必要があるとされる (これらのリード総数はゲノム上に対応付け後、品質管理のための各種フィルタリング後に残ったリードのみを算出対象としている)。 シーケンス深度が十分であるか否かは飽和解析を行うことで実験的に判断することができる。 シーケンス深度が不十分である場合にはtechnical replicatesデータどうしを合わせることなどを検討するとよい。

ゲノム上の同一位置に対応付けられるリード(またはリード対)をduplicatesといい、natural duplicatesとPCR duplicatesに区別される。 前者はゲノム上の同一位置に由来するが相異なるDNA断片から得られたものであり、 後者はライブラリ調製時のPCR増幅過程で導入された同一DNA断片由来のコピーである。 すなわちChIP-seq実験結果において前者はシグナルであり、後者はノイズである。 免疫沈降効率が悪く、少量のDNA試料からPCR増幅して調製したライブラリ試料などは後者を多く含んでいる可能性が考えられる。 計算上、実データから両者を区別し後者のみを除外することは困難である。
ライブラリ中のPCR duplicatesの割合が多くなればなるほど、シーケンス深度を十分に確保しても有意義なデータが得られなくなっていくことが分かる。 そこでライブラリの複雑さを反映したduplicates率が品質管理の指標として利用されている(下図)。

duplicatesがPCRに由来するものか否かを判断する手段としてはBAMファイルをゲノムブラウザで可視化するとよい。
duplicates除去はPCR duplicates除去過程として一般的に確立されている。
BAMファイルを座標軸整序後にPicard MarkDuplicatesを適用することでデータファイル中のduplicatesを検出・除去することができる。
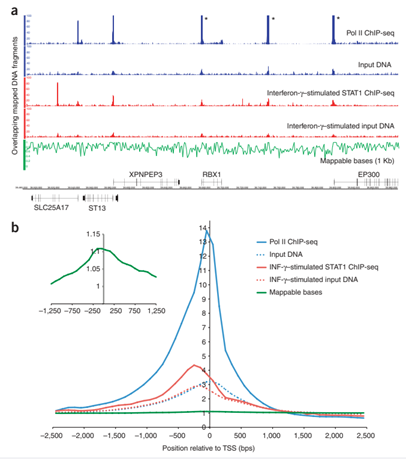
対照試料#のリードがゲノム上で完全にランダムに分布するならば対照試料は必要とされない。 すなわち背景分布として統計分布モデルを想定するだけで本来十分なはずである。 しかしながら実際には上図で端的に例示されているように、ChIP-seq実験には対照試料が必要である。 明らかに対照試料(input DNA)のリードは完全にランダムに分布せず多くの峰を確認することができ、 各峰と一致した位置に目的試料(Pol II免疫沈降試料やSTAT1免疫沈降試料)の峰を確認することができる。 それゆえ目的試料における峰がシグナルであるかノイズであるかの判断には対照試料が必要とされることが分かる。 対照試料が有効な背景分布情報を提供するためには目的試料と同程度以上のシーケンス深度が要求される。
# 対照試料には断片化後DNA(input DNA)やそれをIgGで免疫沈降した試料(IgG control)などが使用される。 IgG controlの方がinput DNAよりも実験操作をより模倣しているが、免疫沈降後のDNA回収量が十分でなければ新たなバイアスを導入する原因となりうる。
ゲノム中にはクロマチンアクセシビリティが他と著明に異なる領域やアライメント不明瞭領域などの偽シグナル源となる特殊領域が知られており、これらも峰検出等を行う前に除去すべきである。 そのような領域の包括的な一覧がENCODEから提供されている ENCODE DAC Exclusion List Regions)。
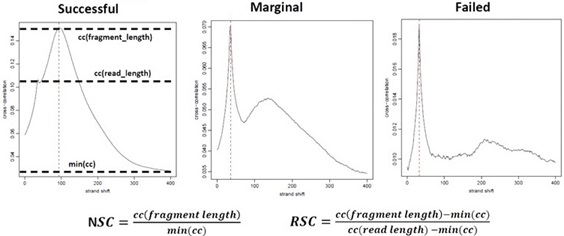
峰検出とは独立して、S/N比を評価することができる代表的な手法にstrand cross-correlationがある。 これは免疫沈降後DNA断片のクラスター形成度を測定する指標であり、次の事実観察に基づいている。
この指標の測定値は正・負鎖間の相互相関、すなわち負鎖をk塩基ずつシフトさせていく過程における正鎖のカバレッジと負鎖のカバレッジとの間におけるピアソン線形相関係数として算出される。
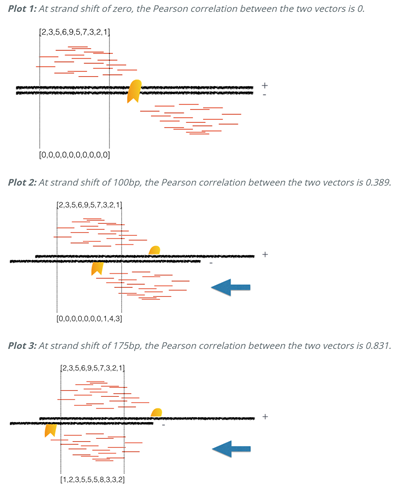
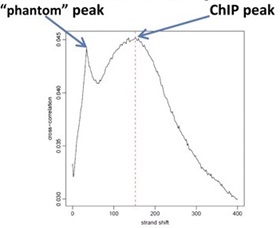
シフト値を横軸方向、係数値を縦軸方向にとりプロットすると、リード長やDNA断片長に対応するシフト位置に峰が形成されるのが一般的である。 断片長位置の係数値と背景係数値との間の比率をNSC(normalized strand cross-correlation coefficient)、 背景係数値で補正後の断片長位置の係数値とリード長位置の係数値との間の比率をRSC(relative strand cross-correlation coefficient)といい、 これらはS/N比を反映する品質指標として利用されている。 NSC>1.05, RSC>0.8が良好な結果の目安とされている。
Irreproducible Discovery Rate(IDR): 順位付けされた峰一覧のペアを比較
はじめの10列はnarrowPeakと同形式であり、2つのreplicateのmerged peaksに関するものである。 7-10列目はランキングに使用したもののみその値が設定され、他は-1に設定される。
Irreproducible Discovery Rate(IDR): 順位付けされたpeak listのペアを比較 replicatesの処理
はじめの10列はnarrowPeakと同形式であり、2つのreplicateのmerged peaksに関するものである。 7-10列目はランキングに使用したもののみその値が設定され、他は-1に設定される。
ゲノム上に対応付けられたリードの領域毎カウントまたは分布形状の情報に基づき峰(ピーク)が定義・検出される。 前者は定義された領域に属するリード数を数え、統計的に有意なリード数を有する領域を峰とする。 後者はリードの空間的分布をモデル化し、リード分布が結合部位近傍の期待分布に適合する場合に峰とする。
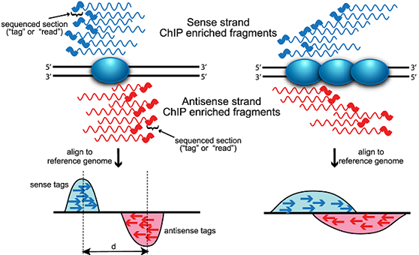
正鎖上の峰と逆鎖上の峰との距離dを定義し、全リードd/2だけ3'端側にシフトすることで正確な結合部位の位置特定を改善する。
峰の対を検出し、シフトモデルを構築するためにmacs2 predictdはデータ全域を走査する。
区間中のfold-enrichmentを-m 5,20のように指定すれば背景分布に対して5-20倍濃縮されたものを峰とする。
macs2 predictd -i xxx.bed -g hs -m 5 20
macs2 callpeakはduplication除去、断片長予測、ピークコールの全てを実行するラッパー関数である。
# For help: macs2 callpeak -h # For ChIP-seq macs2 callpeak \ -t in.bam \ -g hs \ -p 1e-5 \ -f BAM \ -B \ -n prefix \ --outdir outdir # 既定ではnarrow peakを検出する。 # broad peaksを検出する場合は--broadを付与する。 # コントロール試料がある場合は-c CONTROL.bamを付与する。 # -g 生物種(mm|hs) # -B: bigbed出力 # For ATAC-seq macs2 callpeak \ -t in.bam \ -g hs \ -p 1e-5 \ -f BAM \ -B \ -n prefix \ --outdir outdir \ --nomodel \ --shift -read_length \ --extsize 2xread_length # リード長50bpなら--nomodel --shift -50 --extsize 100を追加する。
別章で述べたように範囲データの操作にはbedtoolsを利用する。
# AのうちBと重なるものを標準出力する bedtools intersect -u -a A.narrowPeak -b B.narrowPeak # AのうちBと重ならないものを標準出力する bedtools intersect -v -a A.narrowPeak -b B.narrowPeak
IgGで可視化するなどの目的のためにBAM-BIGWIG変換を行う。 BAMファイルは座標軸で整序され、索引付けされたものである必要がある。
# BAM/BIGWIG変換 (bamCoverage from deeptools)
bamCoverage -b in.bam \
-o out.bw \
-of bigwig \
--normalizeUsing CPM
deeptoolsを利用しTSSなどの周りのピーク分布を確認する。
# 前処理/ TSS周囲のピーク分布を調べる場合
computeMatrix scale-regions \
--regionsFileName genome.gtf \
--scoreFileName xxx.bw \
--outFileName xxx.gz \
--upstream 1000 \
--downstream 1000 \
--skipZeros
# 前処理/ 参照頂点を指定しその周囲のピーク分布を調べる場合
computeMatrix reference-point \
--referencePoint center \
--regionsFileName refPoint.summits.bed \
--scoreFileName xxx.bw \
--outFileName xxx.gz \
--upstream 1000 \
--downstream 1000 \
--skipZeros
# Metagene Plot
plotProfile -m xxx.gz -out xxx.pdf
# Heatmap
plotHeatmap -m xxx.gz -out xxx.pdf
ROSEを利用してSuperEnhancer検出を実行する。 cmhadsoシステムの参照項目(-r)からROSE.1.tar.gzを検索・入手・展開する。 また依存ソフトウェア(samtools, R version > 3.4, bedtools > 2, Python2) を導入する。 入力データであるBAMファイル及びピーク情報ファイル(MACS2などの出力: xxx_peaks.narrowPeak)をROSE.1配下に置く。 両ファイルとも染色体名は"chr"から始まる必要がある。 前準備として、peak2gff.1.shスクリプトを編集・実行しピーク情報をROSEプログラムの入力ファイル形式に合わせる。
# peak2gff.1.shスクリプト
INF=xxx_peaks.narrowPeak
OUTF=xxx_peaks.gff
cat $INF |
awk 'BEGIN{FS=OFS="\t"}{
print $1,$4,"*",$2,$3,$5,$6,0,$4
}' > $OUTF
現状のファイル構成は以下の通りである。
ROSE.1 ├── ROSE_main.py ├── ROSE_bamToGFF.py ├── ROSE_callSuper.R ├── ROSE_utils.py ├── annotation │ ├── README │ ├── hg38_refseq.ucsc │ ├── hg38_refseq_select.ucsc │ ├── mm39_refseq.ucsc │ └── mm39_refseq_select.ucsc ├── peak2gff.1.sh ├── ROSE.1.sh ├── xxx.bam ├── xxx_peaks.narrowPeak └── xxx_peaks.gff
ROSE.1.shスクリプトからROSEプログラムを実行する。
# ROSE.1.sh: ROSEプログラム実行スクリプト # Required software # samtools # R version > 3.4 # bedtools > 2 # python2 # INPUT/OUTPUT INPUT_CONSTITUENT_GFF=xxx_peaks.gff RANKING_BAM=xxx.bam OUTPUT_DIRECTORY=xxx #CONTROL_BAM=xxx.bam # if needed # Annotatipn GENOME_BUILD=mm39s # mm39: mm39_refseq.ucsc # mm39s:mm39_refseq_select.ucsc # hg38: hg38_refseq.ucsc # hg38s:hg38_refseq_select.ucsc # パラメータ STITCHING_DISTANCE=12500 TSS_EXCLUSION_ZONE_SIZE=2500 # 実行 python ROSE.py \ -g $GENOME_BUILD \ -i $INPUT_CONSTITUENT_GFF \ -r $RANKING_BAM \ -o $OUTPUT_DIRECTORY \ -s $STITCHING_DISTANCE \ -t $TSS_EXCLUSION_ZONE_SIZE # -c $CONTROL_BAM # if needed
xxx_peaks_AllEnhancers.table.txtが主要な出力データである。 その他、N_peaks_12KB_STITCHED_TSS_DISTAL_ENHANCER_REGION_MAP.txt, xxx_peaks_SuperEnhancers.table.txt, xxx_peaks_Enhancers_withSuper.bedなどはこれを抽出することで得られるため不要である。 タブ区切り9列構成(1:SE (stitched enhancer)ID, 2:chr, 3:SE start, 4:SE end, 5:stitchedされた構成エンハンサー数, 6:constituents size? , 7:N.BAM?, 8:enhancer rank, 9:isSuper)である。
MEME suiteは結合モチーフ解析ツールの一式であり、新規モチーフ探索にはMEMEやSTREME、モチーフエンリッチメント解析にはSEA、モチーフ走査にはFIMO、モチーフ比較にはTomTom、これらの統合解析にはXSTREMEやMEME-ChIPという専用ツールが提供されている。 MEME suiteではMEME形式( 仕様および 実例 )でモチーフデータの入出力を行う。
SEA (Simple Enrichment Analysis)は調査対象の塩基配列中に、どんな既知モチーフが多く含まれているかを調べるツールである。
sea --text --p input.fa --m MOTIF1.meme --m MOTIF2.meme ... > output.tsv
出力データはタブ区切りの17列(1:RANK, 2:DB, 3:ID, 4:ALT_ID, 5:CONSENSUS, 6:TP, 7:TP%, 8:FP, 9:FP%, 10:ENR_RATIO, 11:SCORE_THR, 12:PVALUE, 13:LOG_PVALUE, 14:EVALUE, 15:LOG_EVALUE, 16:QVALUE, 17:LOG_QVALUE)からなり、例えば以下のようにして必要な情報を取り出すとよい。
cat output.tsv |
grep . |
grep -v '#' |
awk 'BEGIN{FS=OFS="\t"}{
print $1, $3, $4, $16, $5
}'
ヒトやマウスにおける既知モチーフデータはCMHADSOシステムの参照データ(HOCOMOCO_12.tar.gzやJASPAR2024.tar.gz)から入手することができる。 その他の生物種やバージョンのものについてはJASPARやHOCOMOCOのWEBページから入手するとよい。
STREMEは調査対象の塩基配列中で頻出するモチーフを新たに発見するためのツールである。
streme --dna --text --p input.fa > output
FIMO (Find Individual Motif Occurrences)は塩基配列を走査し、既知モチーフに対して個々マッチするか調べるツールである。
Tomtomは調査対象のモチーフ群を既知のモチーフ群と比較し、順位付けするためのツールである。
fimo --oc OUTDIR MOTIF1.meme input.fa
tomtom --text qry.meme MOTIF1.meme MOTIF2.meme ...
ChIPpeakAnnoを利用する。
# 入力データ
gr <- toGRanges("xxx.narrowPeak", format = "narrowPeak", header = F)
# 遺伝子モデル
BiocManager::install("TxDb.....")
library(TxDb....)
gr_ann <- toGRanges(TxDb....)
seqlevels(gr) <- seqlevelsStyle(gr_ann)
# 峰を最近接TSSに対応させる
out <- annotatePeakInBatch(gr, AnnotationData = gr_ann)
# Pie chart
pie(table(out$insideFeature))
# 峰重なり領域の探索
ol <- findOverlapsOfPeaks(gr1, gr2)
makeVennDiagram(ol, NameOfPeaks = C("A", "B"))
overlaps <- ol$peaklist[["gr1", "gr2"]]
out <- assignChrmosomeRegion(
overlaps,
nucleotideLevel=F,
precedence = c(
"Promoters",
"immediate Downstream",
"fiveUTRs",
"threeUTRs",
"Exons",
"Introns"),
TxDb = TxDb....)
pie(out$percentage)
ピーク座標(xxx.narrowPeakなど)をBED形式に変換し、GREATの入力データとする。 ピークに対する遺伝子の割付けはTwo nearest genesを選択すればよい。
CMHADSOシステムの参照データ(chipatlas_20240806.tar.gz)
Lander-Waterman式[PMID: 3294162]: カバレッジの重複度の理論値はLN/G (Lはリード長、Nはリード数、Gはパプロイドゲノム長)
ゲノム上においてリードを一意的に対応づけ可能な配列の割合をmappabilityという。 当然リードが長いほどmappabilityが増加する。