2024年現在シングルセル発現解析の流れとしては、10x Genomics社のシステムを利用してライブラリを作成し、シーケンス後のデータを同社のCell Rangerで一次解析後、Seuratなどのツールを利用して目的に応じた二次解析を行うのが一般的である。 本章ではシングルセル発現解析における一般的なデータ解析例を略述する。
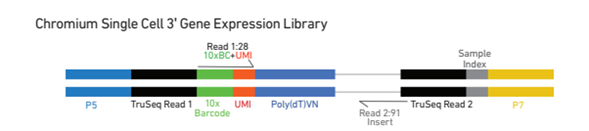
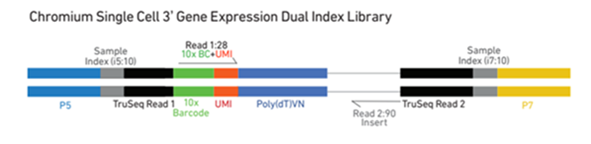
シングルセル発現解析ライブラリ(上図)をシーケンスすることで以下の情報を有するデータ(FASTQファイル)が得られる。
まずはCell Rangerソフトウェアやヒト・マウスのpre-buildリファレンスデータを Downloadsサイトから入手し、 Installガイドに従い、ソフトウェアをインストールする。
学内の計算機サーバーを利用する場合は以上の作業は不要である。 ヒトとマウスのリファレンスデータは共有directoryに配置済であり (ヒト:/home/database/cellranger/refdata-gex-GRCh38-2020-A, マウス: /home/database/cellranger/refdata-gex-mm10-2020-A)、 Cell Rangerは以下のコマンドでロードすることができる。
module load cellrangerヒトやマウス以外のリファレンスが必要な場合などは cellanger mkrefコマンドにより、カスタムリファレンスを作成する。
# === custom reference 作製スクリプト === # 事前準備: Ensembl等からGTFファイルとGENOME配列ファイルを取得し解凍しておく # 1: input/outputデータ inputGTF=xxxx.gtf # 事前入手・解凍したGTFファイル inputGENOME=xxxx.fa # 事前入手・解凍したGENOMEファイル filteredGTF=xxxx.gtf # 中間出力結果 outputDir=xxxx # この出力結果をcellranger countのreferenceに使用する # 2: GTFファイルからprotein_codingのエントリを抽出 cellranger mkgtf \ $inputGTF \ $filteredGTF \ --attribute=gene_biotype:protein_coding # 3: Reference作製 cellranger mkref \ --genome=$outputDir \ --fasta=$inputGENOME \ --genes=$filteredGTF
入力データであるFASTQファイルは以下のどちらかの命名規則に従う必要がある。
readTypeはI1, I2, R1, R2(上述の10x libraryの項目参照)のいずれかであり、I1,I2のファイルは存在しなくてもよい。
次のような入力データを想定した場合、以下の解析スクリプトにおいてinputDirにSRC, inputIDにはdata1, data2, data3を順次指定していけばよい #。
#解析に慣れてきたら下記スクリプトにおいてdata1を$1に変えるなどして自分なりの改変を加えること! 本章の以降のスクリプト例でも同様。
# 入力データ例
SRC
├── data1
│ ├── data1_S1_L004_I1_001.fastq.gz
│ ├── data1_S1_L004_R1_001.fastq.gz
│ └── data1_S1_L004_R2_001.fastq.gz
├── data2
│ ├── data2_S1_L005_I1_001.fastq.gz
│ ├── data2_S1_L005_R1_001.fastq.gz
│ └── data2_S1_L005_R2_001.fastq.gz
└── data3
├── data3_S1_L006_I1_001.fastq.gz
├── data3_S1_L006_R1_001.fastq.gz
└── data3_S1_L006_R2_001.fastq.gz
# === cellranger count 実行スクリプト===
# 1: input/output Directory
inputDir=SRC # inputの最上位directory名
inputID=data1 # その配下の各試料を区別するdirectory
# --fastqオプションで指定するもの
# さらにその配下にFASTQデータを置く
outputID=data1 # output directory名 (inputIDと同名でよい)
# 2: Reference
reference=/home/database/cellranger/refdata-gex-mm10-2020-A # mouse
#reference=/home/database/cellranger/refdata-gex-GRCh38-2020-A # human
# 3: CPU/Memory
localcores=10 # 使用するCPU数
localmem=96 # 使用するメモリ数
# 4: cellranger countの実行
cellranger count \
--id=$outputID \
--fastqs=$inputDir/$inputID \
--transcriptome=$reference \
--localcores=$localcores \
--localmem=$localmem
必要に応じてSoupXによるambient RNA除去を検討するとよい。 SoupXの入力データとしてcellranger countの出力結果であるfiltered_feature_bc_matrixおよびraw_feature_bc_matrixを格納したdirectoryを準備する。 SoupXには細胞クラスタリング済みの入力データを与えることが望ましいことから、Seuratなどを利用して事前に細胞クラスタリングを実行しておく。
次のような入力データを想定した場合のスクリプトの具体例を示す。
# 入力データ data ├── filtered_feature_bc_matrix │ ├── barcodes.tsv.gz │ ├── features.tsv.gz │ └── matrix.mtx.gz └── raw_feature_bc_matrix ├── barcodes.tsv.gz ├── features.tsv.gz └── matrix.mtx.gz
# === SoupX 実行スクリプト========================
# 必要なソフトウェア: SoupX, Seurat, DropletUtils
# ================================================
# 1: ライブラリの読込み
library("SoupX")
library("Seurat")
library("DropletUtils")
# 2: Seuratによる細胞クラスタリング
so <- CreateSeuratObject(Read10X("data/filtered_feature_bc_matrix"))
so <- NormalizeData(so)
so <- FindVariableFeatures(so)
so <- ScaleData(so)
so <- RunPCA(so)
so <- RunUMAP(so)
so <- FindNeighbors(so)
so <- FindClusters(so)
# 3: SoupXによるカウント数補正
sc <- load10X("data")
sc <- setClusters(sc, setNames(so$seurat_clusters, colnames(so)))
sc <- autoEstCont(sc)
sc <- adjustCounts(sc)
# 5: DropletUtilsによる結果出力
# 以後これを"filtered_feature_bc_matrix"の代用とする
DropletUtils::write10xCounts("outputDir", sc)
Tutorials| Cheat Sheet| API| FAQ
Libraryを読込む。
library(Seurat) library(ggplot2) # ggsave()関数を使用する際に必要
CELLRANGERの出力結果を入力としSeurat Objectを作る。
Read10X()関数にCellRanger出力データ(filtered_feature_bc_matrix#1)を与えることでdgCmatrixクラスの疎行列#2が得られる。 さらにこれを CreateSeuratObject()関数に与えることで、Seurat Object (以下soと略称する)を作製することができる#3。 カウントデータはdgCmatrixクラスの疎行列としてso@assays$RNA$countsに格納されている。 メタデータの初期値としてso@meta.data$orig.ident, so@meta.data$nCount_RNA (細胞ごとのUMI数), so@meta.data$nFeature_RNA (細胞ごとの検出遺伝子数)が設定されている。
#1
このdirectory中には3種のデータが含まれており、barcodes.tsv.gzは細胞識別用バーコード、
features.tsv.gzは遺伝子名情報、matrix.mtx.gzはMatrix Market形式(疎行列)で記述されたカウントデータである。
#2
as.matrix()関数により疎行列から通常の行列に変換することができる。
#3
min.cellsやmin.features引数を指定すれば、オブジェクト作成時にそれぞれ遺伝子や細胞のフィルタリングを行うことができる。
inputDir = "filtered_feature_bc_matrix" so <- CreateSeuratObject(Read10X(inputDir))
次にデータ品質指標を確認し必要に応じてフィルタリングを行う。
# ミトコンドリア百分率情報をメタデータに追加
so[["percent.mt"]] <- PercentageFeatureSet(so, pattern="^mt-") # ヒトの場合マッチパターンは"^MT-"
# 可視化
VlnPlot(so, features=c("nFeature_RNA", "nCount_RNA", "percent.mt"))
# 可視化結果を踏まえてパラメータ値を定めフィルタリング実行
so <- subset(so, subset=nCount_RNA > 1000 & percent.mt < 20)
# 再度可視化し、フィルタリング実行結果を確認
VlnPlot(so, features=c("nFeature_RNA", "nCount_RNA", "percent.mt"))
次にデータ正規化・発現変動遺伝子の同定・スケール化を実行する。
NormalizeData()関数の実行結果はso@assays$RNA@dataに出力される。
FindVariableFeatures()関数により顕著に発現変動している遺伝子(features)を同定する。 既定では発現変動遺伝子の選択法はselection.method="vst"、選択遺伝子数はnfeatures=2000 に設定されている。 関数実行結果はso@assays$RNA@var.features, so@assay$RNA@meta.featuresに出力される。
ScaleData()関数は so@assays$RNA@dataをscale(拡大縮小)/center(中央配置)したものをso@assay$RNA@scale.dataに出力する。 既定ではvariable featuresのみがスケール化対象であるが、features=rownames(so)のように引数指定すると全遺伝子が対象となる#。
vars.to.regressで引数指定された場合、その変数値に対する回帰で得られた残差をスケール化したもの返すこともできる。
# もちろん両者とも算出値自体は同一であるが、後者の場合には解析対象遺伝子数が多いため、解析時間及びデータ量が著明に増加することに注意が必要である。
so <- NormalizeData(so) so <- FindVariableFeatures(so) so <- ScaleData(so)
次元削減・最近傍グラフ構築・クラスター検出を実行する。
RunPCA()関数は主成分分析による次元削減を行う関数で、その実行結果はso@reductions$pcaに設定される。 既定では現在のAssayにおけるvariable features (スケール化データ中に含まれている必要がある)を使用する。
FindNeighbors()関数によりk最近傍グラフを構築する。 その実行結果はso@graphs$RNA_nn, so@graphs$RNA_snnに設定される。
FindClusters()関数 により共有最近傍に基づくクラスタリングを実施する。 その実行結果はso@meta.data$RNA_snn_res.N, so@meta.data$seurat_clustersに設定される。
RunUMAP()関数の実行結果、so@reductions$umapが設定される。
so <- RunPCA(so, verbose = F) so <- FindNeighbors(so, dims = 1:20) so <- FindClusters(so, resolution = 0.5) so <- RunUMAP(so, dims = 1:20, verbose = F)
2次元上での細胞クラスターの分布や遺伝子発現量を視覚化する。
DimPlot(so, reduction = "umap")
VlnPlot(so, features = c("gene"))
FeaturePlot(so, features = c("gene"))
オブジェクトの保存・読込はRの一般的なやり方に従う。
saveRDS(so, "so.rds") # 保存
so <- readRDS("so.rds") # 読込
# soの全体構造の関する情報
dim(so) # FxC: Fは遺伝子数, Cは細胞数
str(so, max.level=2)
# アクセス・サブセット化(データフレームと同要領)
so[f, c] # data.frameと同様。f, cはベクトル
rownames(so) # 遺伝子名
colnames(so) # 細胞名, Cells(so)というアクセサ関数も同様の出力結果.
# Idents
Idents(so) # so@active.identでもよい。名前付きファクター出力
# 名前: 細胞名
# 値: クラスター名・細胞種名
Idents(so) >- vec # idents更新。自動でベクターがファクターに変換される。
# メタ情報
so@meta.data # CxL型のデータフレーム: Cは細胞数, Lは列数
so[[]] # 簡略記法
# 対応関係の確認
all(names(Idents(so)) == colnames(so)) # 結果: TRUE
all(names(Idents(so)) == rownames(so[[]])) # 結果: TRUE
all(names(Idents(so)) == Cells(so)) # 結果: TRUE
# 発現値データの確認
so@assays$RNA@counts # 生のカウントデータ
so@assays$RNA@data # ライブラリサイズで正規化およびlog変換済みデータ
so@assays$RNA@scale.data # scale化済みデータ
以上を踏まえれば、例えば PercentageFeatureSet(so, pattern="^MT-")に相当する処理は以下のように実行できることが分かる。
mt = grep("^MT-", rownames(so))
a = apply(so@assays$RNA@counts, 2, sum)
b = apply(so[mt, ]@assays$RNA@counts, 2, sum)
b / a * 100
また細胞種ごとのデータを個別取得するには以下のようにする。
so[, Idents(so) == "cellType1"] # 関数を用いてsubset(so, idents=c("cellType1"))ともできる。
so$NEW <- vec # vector代入
メタデータ追加を挙げる。 患者番号(so$P)の情報をもとに患者型(so$PType, その値はAかBとする)を新たに導入するには以下のようにする。
A <- c("S1", "S3", "S5")
B <- c("S2", "S4", "S6")
so@meta.data$PType[so$P %in% A] <- "A"
so@meta.data$PType[so$P %in% B] <- "B"
so$PType <- factor(so$PType, levels=c("A", "B"))
1:1型のRename例を下に示す。
map1to1 = data.frame(
fr = c("B cells", "T cells"),
to = c("B", "T")
)
for(i in 1:nrow(map1to1)){
so$cellType[id == map1to1[i, 1]] = map1to1[i, 2]
}
Idents(so) = sub('old', 'new', Idents(so)
# Identsの名前更新については専用の関数を利用することもできる
so <- RenameIdents(so, 'old' = 'new')
まずはサブクラスターの細胞種情報(subID)を抽出する。
# Get subIDs subID1 <- Idents(sub_so1) subID2 <- Idents(sub_so2) : :
また元データの細胞種情報(soID)をメタデータにコピーする。
# idents to meta soID <- Idents(so) so$NEW <- as.vector(soID)
名前付きベクターによる1:N型のRenameを実行する。
# 1> subIDリストを作る
subIDList <- list(subID1, subID2, ...)
# 2> Convert named factor to named vector
# (as.vectorは名前属性を消すので先に退避しておく)
subIDList = lapply(subIDList, function(fct){
name = names(fct)
vec = as.vector(fct)
names(vec) = name
return(vec)
}
)
# 3> Rename by named vector
for(i in 1:length(subIDList)){
so$NEW <- subIDList[[i]]
}
# 4> meta to idents
Idents(so) <- so$NEW
細胞クラスター・細胞種分布をUMAP上などの2次元空間上で視覚化するには
# 基本形 DimPlot(so, reduction = "umap") # 見た目の微調整 DimPlot(so, reduction = "umap", label = TRUE, pt.size = .8, label.size = 8)
遺伝子発現をバイオリンプロットで視覚化するには
# 基本形 (identsごとの発現量)
VlnPlot(so, features = c("gene"))
# 群間比較
VlnPlot(so, features = c("gene"), split.by = "column_name" , split.plot = T)
遺伝子発現をUMAP上などの2次元空間上で視覚化するには
FeaturePlot(so, features = c("gene"))
個々の遺伝子の代わりに遺伝子セットレベルに集約された発現量をUMAP上などの2次元空間上で視覚化するには
library(UCell)
# 細胞種ごとのgenesetをlistとして定義
GSL <- list()
GSL$cellType1 <- c("gene1_1", "gene1_2", ....)
GSL$cellType2 <- c("gene2_1", "gene2_2", ....)
GSL$cellType3 <- c("gene3_1", "gene3_2", ....)
# 発現情報の集約
so <- AddModuleScore_UCell(so, features = GSL, name = NULL)
so <- SmoothKNN(so, signature.names = names(GSL), reduction = "pca")
# 視覚化
FeaturePlot(so, features = names(GSL))
ドットプロットによる視覚化をするには
# 細胞種の並び順を指定(縦軸方向)
cellTypeOrder <- c("cellType", .....)
Idents(so) <- factor(Idents(so), levels = rev(cellTypeOrder)) # rev()で指定順を反転させていることに注意
# 遺伝子の並び順を指定(横軸方向)
GENEs <- c("gene", ....)
GENEs <- factor(GENEs, levels = GENEs)
# 視覚化
DotPlot(so, features = GENEs, assay = "RNA") + RotatedAxis()
各細胞種の割合の群間比較を表す棒グラフ表示するには
# definition of graph component: Color Pallete
CP = c("#nnnnnn", "#nnnnnn", ...)
# data (X1(細胞種情報): 縦方向の分割 X2(群間比較情報): X軸方向のvs)
df <- as.data.frame(table(so$X1, so$X2))
colnames(df) <- c("X1", "X2", "V")
# Plot
ggplot(df) +
geom_bar(aes(X2, V, fill = X1),
stat = "identity",
position = "fill",
linewidth = .5,
color = "black",
width = .8
) +
# -------------------------------------------------------
scale_y_continuous(expand=c(0, 0)) +
ylab("fraction") +
theme_classic() +
theme(
legend.position = "none",
text = element_text(size = 18, family="Helvetica"),
axis.title.x = element_blank(),
axis.text.x = element_text(angle = 30, hjust = 1,vjust = 1)
) +
scale_fill_manual(values = CP, limits = rev(levels(df$X1))) # + coord_flip()
# -------------------------------------------------------
# ggsave("xxx.png", height = xxx, width = xxx)
UMAPグラフ表示を制御するには
# Color Palette
CP = c("#nnnnnn", "#nnnnnn", ...)
# Get Data
tmp <- DimPlot(so, reduction="umap")
df <- tmp$data
# add Group info
df["GROUP"] <- ..........
# ---------------------------------
# Plot: Patient Origin
# umap_1, umap_2 may be UMAP_1, UMAP2!!
ggplot(df, aes(umap_1, umap_2, color = GROUP)) +
geom_point(
size = .3,
stroke = .3,
shape = 16
) +
# -------------------------
# this is for point size of legend
geom_point(
data = df[df["GROUP"]=="NEVER_MATCHED",],
aes(umap_1, umap_2, color = GROUP),
size = 3,
stroke = 2,
shape = 16,
) +
# -------------------------
scale_color_manual(name = "",
values=CP,
labels=c(
"Type1 (N = 4,424)",
"Type2 (N = 5,994)",
...
)
) +
theme_classic() +
theme(
text = element_text(size=18, family="Helvetica"),
axis.text.x = element_blank(),
axis.text.y = element_blank(),
axis.ticks.x = element_blank(),
axis.ticks.y = element_blank()
) +
xlab("UMAP1") +
ylab("UMAP2") +
# title
ggtitle("xxxxx") +
theme(
plot.title = element_text(hjust=.5)
)
各クラスターのマーカー遺伝子を検出するには
DEGs <- FindAllMarkers(so, verbose = FALSE)
DEGs_pos <- DEGs[DEGs$avg_log2FC > 0,]
DEGs_neg <- DEGs[DEGs$avg_log2FC < 0,]
top10 <- do.call(rbind, lapply(split(DEGs_pos, DEGs_pos$cluster), function(x) {
x[order(x$avg_log2FC, decreasing = TRUE), ][1:10,]}))
top10 <- na.omit(top10)
DEGs = DEGs[c(7, 6, 2, 5, 1, 3, 4)]
# 1: p_val 2: avg_log2FC 3: pct.1 4: pct:2 5: p_val_adj 6: cluster 7: gene
write.table(DEGs, "xxx.csv", sep = ",", row.names = F)
#@ DEGsのHeatmap図示及び保存
DoHeatmap(so, top10$gene)
table(Idents(so)) table(Idents(so), so$PType)
conda install -c conda-forge r-remotes
remotes::install_github('chris-mcginnis-ucsf/DoubletFinder')
library(Seurat) require(DoubletFinder)
以下のデータはprefiltering, scaling, PCA, UMAP等を解析済みもの. Aggregateしたデータは使用しない (ex. Ctrlデータ + 介入データ)
so <- readRDS("tmp_a.rds")
so_tmp_1 <- paramSweep_v3(so, PCs = 1:16) so_tmp_2 <- summarizeSweep(so_tmp_1, GT = FALSE) bcmvn <- find.pK(so_tmp_2) barplot(bcmvn$BCmetric, names.arg=bcmvn$pK,las = 2) # Bcmvn ~ Mean-variance normalized bimodality coefficient # 下図のMax値である0.02を次式のpKに設定
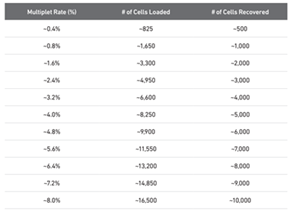
# Set Doublets numbers
nExp <- round(ncol(so) * 0.10)
# doubletsを10%と仮定
# 10x社の細胞インプット数と回収数の対応表を参照して決定
so <- doubletFinder_v3(so,
pN = 0.25,
pK = 0.02,
nExp = nExp,
PCs = 1:16)
# PCs ~ The number of statistically-significant principal components
#
# pN ~ This defines the number of generated artificial doublets,
# expressed as a proportion of the merged real-artificial data.
# Default is set to 25%, based on observation that
# DoubletFinder performance is largely pN-invariant
#
# pK ~ This defines the PC neighborhood size used to compute pANN,
# expressed as a proportion of the merged real-artificial data
# No default is set, as pK should be adjusted for each scRNA-seq dataset.
#
# nExp ~ This defines the pANN threshold used to make final doublet/singlet predictions.
DF.name = colnames(so@meta.data)[grepl("DF.classification", colnames(so@meta.data))]
DimPlot(so, group.by = DF.name, pt.size = 1.2)
VlnPlot(so,features = "nFeature_RNA", group.by = DF.name, pt.size = 1.2)
so_filtered = so[,so@meta.data[,DF.name] == "Singlet"]
# 事前準備: 各データの発現変動遺伝子を取得しておく so_1 <- NormalizeData(so_1) so_2 <- NormalizeData(so_2) so_1 <- FindVariableFeatures(so_1) so_2 <- FindVariableFeatures(so_2) # 統合実行: Seurat objectのリストを入力として与える so <- IntegrateData(FindIntegrationAnchors(c(so_1, so_2))) # DefaultAssayを"integrated"に設定した状態でScaleData以後の # クラスタリング手順を実行していく
sub <- subset(so, idents=c("cellType1"))
データの再統合を実施する場合と実施しない場合の例を以下に挙げる。 FindVariableFeatures()およびScaleData()は再実行することが望ましいが( #1883, #6115, #2365 )、 NormalizeData()は再実行する必要はない( #678)。
# データの再統合を実施する場合
DefaultAssay(sub) <- "RNA"
tmps <- SplitObject(sub, split.by = "xxxx")
tmps <- lapply(tmps, function(x){
x <- FindVariableFeatures(x)
})
SUB <- IntegrateData(FindIntegrationAnchors(tmps))
DefaultAssay(SUB) <- "RNA"
SUB <- ScaleData(SUB, features = rownames(SUB))
DefaultAssay(SUB) <- "integrated"
SUB <- ScaleData(SUB)
SUB <- RunPCA(SUB)
SUB <- FindNeighbors(SUB)
SUB <- FindClusters(SUB)
SUB <- RunUMAP(SUB)
# データの再統合を実施しない場合 DefaultAssay(sub) <- "RNA" sub <- ScaleData(sub) sub <- FindVariableFeatures(sub) DefaultAssay(sub) <- "integrated" so <- ScaleData(sub) sub <- RunPCA(sub) sub <- FindNeighbors(sub) sub <- FindClusters(sub) sub <- RunUMAP(sub)
以上の例ではAssayがRNAの場合にもScaleDataを実行しているが後にヒートマップの作製などに利用することを想定しているためであり、必ずしも必要な操作ではない。
In the folling example, Test data is Seurat object and reference data is SingleCellExperiment object. Cell annotation is stocked at LABEL column of reference data.
library(SingleR) library(SingleCellExperiment) library(Seurat)
so <- readRDS("SO.rds")
sceT <- as.SingleCellExperiment(so)
sceR <- readRDS("SCE.rds")
pred <- SingleR(
sceT,
sceR,
sceR$LABEL,
assay.type.test = 1
)
so$pred <- pred$labels
In the folling example, Test data is Seurat object and reference data is SingleCellExperiment object. Cell annotation is stocked at LABEL column of reference data.
library(SingleR) library(SingleCellExperiment) library(Seurat) library(scran)
so <- readRDS("SO.rds")
sceT <- as.SingleCellExperiment(so)
sceR <- readRDS("SCE.rds")
pred <- SingleR(
sceT,
sceR,
sceR$LABEL,
de.method = "wilcox"
de.n = 50
)
so$pred <- pred$labels
head(sort(table(pred$labels), decreasing = TRUE))
to.remove <- is.na(pred$pruned.labels) table(Label = pred$labels, Removed = to.remove)
to.remove <- pruneScores(pred, min.diff.med = 0.2) table(Label = pred$labels, Removed = to.remove)
plotScoreHeatmap(pred)
plotDeltaDistribution(pred)
meta data include cell annotation LABEL.
library(SingleCellExperiment) library(scuttle) # logNormCounts()
count <- read.table("CountData")
meta <- read.table("MetaData")
sce <- SingleCellExperiment( assays = list( counts = as.matrix (count)), colData = meta ) # Remove cells that do not belong to any Clusters (if needed) # For example: sce <- sce[,sce$LABEL != ""]
# IMPORTANT!: For SingleR, raw counts MUST be converted to logNorm data sce <- logNormCounts(sce)
saveRDS(sce, "SCE.rds")
インストールガイドに従い、各種ソフトウェアをインストールする。 以下、解析手順を略述するが、詳細・最新情報についてはチュートリアル 1, 2, 3を参照にすること。
ライブラリとデータ(Seurat Object)を読込み、Seurat-CellChatオブジェクト変換関数("so2cc")を適用する。
# ライブラリ読込み suppressPackageStartupMessages(library(CellChat)) suppressPackageStartupMessages(library(Seurat)) suppressPackageStartupMessages(library(ggplot2)) suppressPackageStartupMessages(library(ComplexHeatmap))
# データ(Seurat Object)読込み
so <- readRDS("xxx.rds")
# 変換関数の定義
so2cc <- function(
so,
split.by = NULL,
assay = "RNA",
slot = "data",
animal = c("human","mouse"),
min.cells = 10,
lift = FALSE
){
# This function convert integrated seurat object with multiple conditions
# to a list of Cellchat objects, following by basic processing.
# validate the args
if(is.null(split.by)) stop("split.by is not properly set")
if(!any(split.by == names(so[[]]))) stop("split.by is not properly set")
if(!(animal %in% c("human", "mouse"))) stop("split.by is not properly set")
if(!is.logical(lift)) stop("split.by is not properly set")
# all groups (used for liftCellChat function)
allGroup <- unique(Idents(so))
# convert to cellchat object
L <- SplitObject(so, split.by = split.by)
cc <- list()
for(i in 1:length(L)){
data <- GetAssayData(L[[i]], assay = assay, slot = slot)
labels <- Idents(L[[i]])
meta <- data.frame(labels = labels, row.names = names(labels))
cellchat <- createCellChat(data, meta = meta, group.by = "labels")
# basic processing of cellchat object
if(animal == "mouse") {
cellchat@DB <- CellChatDB.mouse }
else {
cellchat@DB <- CellChatDB.human }
cellchat <- subsetData(cellchat)
cellchat <- identifyOverExpressedGenes(cellchat)
cellchat <- identifyOverExpressedInteractions(cellchat)
cellchat <- computeCommunProb(cellchat)
cellchat <- filterCommunication(cellchat, min.cells = min.cells)
cellchat <- computeCommunProbPathway(cellchat)
cellchat <- aggregateNet(cellchat)
cellchat <- netAnalysis_computeCentrality(cellchat, slot.name = "netP")
cc[[i]] <- cellchat
# liftCellChat (if needed)
if(lift) cc[[i]] <- liftCellChat(cc[[i]], allGroup)
}
# merge
names(cc) <- names(L)
cc_merged <- mergeCellChat(cc, add.names = names(cc))
# output
return(list(cc_merged, cc))
}
# 変換実行 cc <- so2cc(so, split.by = "xxx", animal = "human", lift=T) # split.byにはsoメタデータの列名を指定(比較対象) # animalにはmouseかhumanを指定 # liftには論理値を指定(細胞種構成が異なるかどうか) # 保存 saveRDS(cc, "xxx.rds")
以下の手順で、細胞間コミュニケーションに関するデータを取得・可視化する。
M <- cc[[1]] # merge形式データを取得する L <- cc[[2]] # list形式データを取得する
まずは引数指定の際に必要となる情報を確認しておく。
names(L) # 試料条件比較順の確認 levels(M@idents$joint) # 細胞種名順の確認
個々のリガンド-レセプター(LR)レベルの比較データは以下例のようにして視覚化できる。
netVisual_bubble(
M,
sources.use = 1, # 1番目の細胞種
targets.use = c(3:5), # 3-5番目の細胞種
comparison = c(1,2), # 2nd vs 1st
remove.isolate = T,
max.dataset = 1, # Higher in 1stのデータを抽出
font.size = 16
)
# ggsave("xxx.png") # ファイル出力する場合
全てのLRデータをExportしたい場合は以下のようにする。
data <- netVisual_bubble( M, comparison = c(1,2), return.data = T ) write.table(data$communication, "LR.csv", sep=",", row.names = F)
以下のスクリプトでデータ形式を変換しておくと比較が容易である。
# parsrc.sh, yarr, delfコマンドをhttps://github.com/ShellShoccar-jpnより入手し使用している
tmp=$(date +%Y%m%d%H%M%S)$$
input=LR.csv
output=LR
# CSVファイルを3列形式に変換
cat $input |
parsrc.sh |
# 必要なデータを抽出
awk '$2~/^(1|2|3|4|5|15)$/' |
#1:source
#2:target
#3:ligand
#4:receptor
#5:prob
#15:dataset
# 3列目の名称のスペースを"_"に変換
tr ' ' '_' |
sed 's/_/ /' |
sed 's/_/ /' |
# 行指向に変換/ header除去
delf 2 |
yarr num=1 |
tail -n +2 |
delf 1 |
# 6/8列形式に変換(後ろの方が大きい値)
sort -k 1,1 -k 2,2 -k 3,3 -k 4,4 -k 5g,5 |
yarr num=4 > $tmp-68
# 6列データ(Not Paired)の処理
cat $tmp-68 |
awk 'NF==6' |
awk '{print $1,$2,$3,$4,$5,$5,$6, "F"}' > $tmp-6
# 8列データ(Paired)の処理
cat $tmp-68 |
awk 'NF==8' |
awk '{print $1,$2,$3,$4,$7,$7-$5,$8, "T"}' > $tmp-8
{
echo "source target ligand receptor prob probDiff dataset isPair"
cat $tmp-6 $tmp-8
} > $output
# 後片付
rm $tmp-68 $tmp-6 $tmp-8
Circle Plotで図示したものをファイル出力する場合は以下のようにする。
png("xxx.png", width=xxx, height=xxx, res=xxx)
# "G = "のように付値の形にしておかないとファイル出力時にエラーが出るので注意
G = netVisual_chord_gene(L[[2]],
sources.use = xxx,
targets.use = xxx,
signaling = "xxx",
lab.cex = xxx)
dev.off()
LR/Signalingデータベース情報は以下のようにしてExportできる。
data <- CellChatDB.mouse
names(data) # interaction, complex, cofactor, geneinfoと出力される
# 主データであるdata$interactionをexportする
write.table(data$interaction, "db.csv", sep=",", row.names = F)
例えばシグナル名とLR遺伝子名の対応表を得るには以下のような形式変換するとよい。 (出力はタブ区切り4列: 1:pathway, 2:signal, 3:ligand, 4:receptor)。
# parsrc.shコマンドをhttps://github.com/ShellShoccar-jpnより入手し使用している
input=db.csv
output=db
# CSVファイルを3列形式に変換後、必要な情報のみ抽出
cat $input |
parsrc.sh |
#1:interaction_name
#2:pathway_name
#3:ligand
#4:receptor
#5:agonist
#6:antagonist
#7:co_A_receptor
#8:co_I_receptor
#9:evidence
#10:annotation
#11:interaction_name_2
awk '$2~/^(2|11)$/' |
awk '
$2==2 {$1=""; $2=""; pathway=$0}
$2==11 {n=$1 ; $1="" ; $2="" ; interact=$0
split(interact, a, "-")
ligand = a[1]
receptor = a[2]
sub("+", ";", receptor)
sub("\\(", "", receptor)
sub("\\)", "", receptor)
# output
printf("%s\t%s\t%s\t%s\n", n, pathway, ligand, receptor)
}' |
{
echo "1 pathway ligand receptor" | tr ' ' '\t'
tail -n +2
} > $output
suppressPackageStartupMessages(library(Seurat)) suppressPackageStartupMessages(library(SeuratWrappers)) suppressPackageStartupMessages(library(monocle3))
so <- readRDS("*.rds")
cds <- as.cell_data_set(so) cds <- estimate_size_factors(cds) cds <- cluster_cells(cds, reduction_method = "UMAP") cds <- learn_graph(cds, use_partition = T)
plot_cells(cds,
color_cells_by = "partition",
label_principal_point = T,
graph_label_size = 6,
cell_size = .5)
plot_cells(cds,
color_cells_by = "partition",
graph_label_size = 6,
cell_size = 1)
cds <- order_cells(cds, root_pr_nodes = c("Y_*"))
plot_cells(cds,
color_cells_by = "pseudotime",
show_trajectory_graph = T,
graph_label_size = 6,
cell_size = 1)
genes <- c("Col1a1")
sub_cds <- cds[row.names(cds) %in% genes,]
plot_genes_in_pseudotime(sub_cds,
label_by_short_name = F,
color_cells_by = "seurat_clusters",
min_expr = 0.1)